
 MENU
MENU 

Precision health in the palm of your hand
Recent breakthrough developments in technologies for real-time genome sequencing, analysis, and diagnosis are poised to deliver a new standard of personalized care.

 Enlarge
EnlargePrecision health is an approach to wellness that takes into account variability in genes, environment, and lifestyle for each person. And thanks to advancements in technology, it’s here today. Huge amounts of data are being collected and analyzed to manage our care, with data sources including laboratory tests, biometric sensors, patient records, hospital data, and more. But results can be slow in coming, and the wait between testing and diagnosis can be days or weeks.
However, recent breakthrough developments in technologies for real-time genome sequencing, analysis, and diagnosis are poised to deliver a new standard of personalized care.
Imagine a case in which a patient is admitted to a clinic and a simple blood or saliva test is administered. Before the visit is over, a complete diagnosis and personalized treatment plan is available. In another scenario, a surgeon who is seeking to remove a tumor with minimal impact to healthy tissue could confirm decisions through real-time tissue sample analysis. Finally, picture a portable pathogen detector that could alert a user to dangerous exposure during a pandemic or disease outbreak.
The key to making these and other visions real would be a handheld device that provides real-time genomic sequencing and analysis of patient DNA or pathogen DNA or RNA.
Advances in genetic sequencing
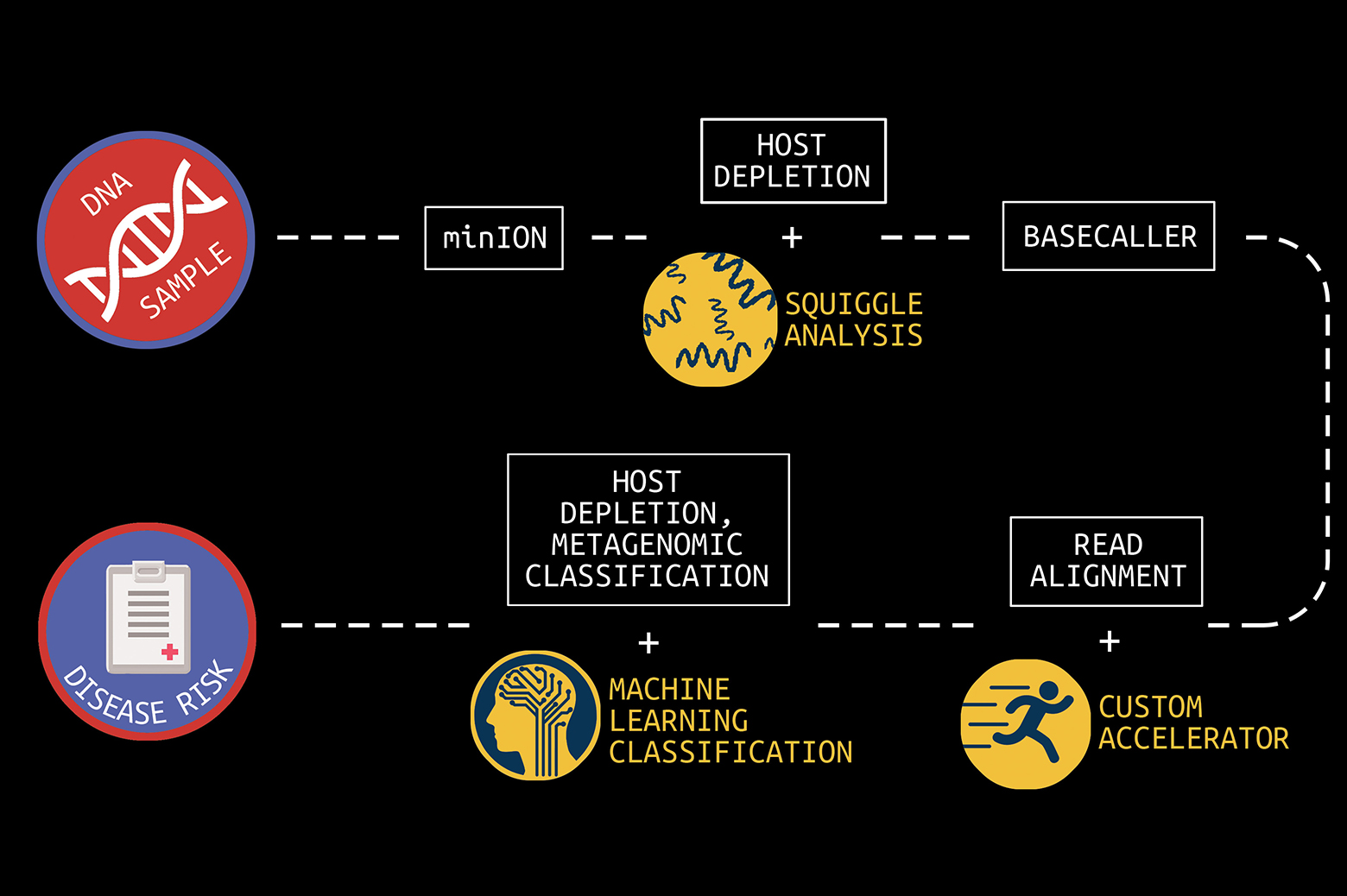
It cost nearly $3 billion to sequence the first human genome in 2001. Today, the cost to sequence a whole human genome is under $1000 and expected to reach about $100 soon. In addition, first- and second-generation sequencing systems were large, expensive, and designed for batch operation. Results would become available days or more after samples were taken. But new, lower-cost third-generation sequencing systems now exist, such as the Oxford Nanopore MinION, which can rapidly sequence individual samples and fit in the palm of your hand.
The human genome is made up of over three billion base pairs of DNA. To sequence a genome, the MinION employs small nanopores to divide a collected sample into billions of strands, called “reads.”
“The MinION is a great handheld sequencing tool and is capable of rapidly sequencing biological data,” says Reetuparna Das, an associate professor in CSE. “It takes the chemical sample, divides the DNA or RNA into strands, and sequences those strands into electrical signals, known as ‘squiggles.’ However, it does not have the compute capability to analyze raw data in the field and quickly produce actionable results.”
All that stands between us and real-time diagnosis is a computing system that can analyze the sequenced data and provide treatment and therapy recommendations before the patient even leaves the office.
The computing challenges
In what is known as secondary analysis, it is the job of a computing system to interpret squiggles as base pairs of DNA, a process which is known as basecalling. A base pair is essentially one rung on a DNA or RNA structure’s ladder. Following that, the system must align the read data to genome reference data and then identify variants between the sample and the reference. The variant data of human genomes is used to identify a genetic disease marker. Sequencing is also used to identify pathogens by aligning DNA or RNA strands to a reference pathogen database and using metagenomic classification tools.
And although this sounds straightforward, sequencing produces about GBs to TBs of data and the processing challenges are steep because of the precision, complexity, and scale of the task. Two multidisciplinary teams of researchers at U-M are working on approaches to overcome this hurdle.
Associate professor Reetuparna Das and professor Satish Narayanasamy, along with professor David Blaauw in Electrical and Computer Engineering, are leading a team funded by the National Science Foundation and the Kahn Foundation that is developing a hardware/software platform to accelerate next-generation genomic sequencing with a focus on pathogen detection and early cancer detection. In this effort, they are collaborating with associate professor of internal medicine and of microbiology and immunology Robert Dickson and assistant professor Carl Koschmann of pediatrics, as well as with associate professor Jenna Wiens, who is also a part of the second research team.
The second team, funded by the Kahn Foundation, is developing data acquisition and machine learning techniques to dramatically improve the prediction, treatment, and management of disease in aging populations. A key component of this effort is the use of machine learning to speed metagenomic analyses.
This large-scale interdisciplinary effort is a collaboration between researchers at Technion – Israel Institute of Technology, the Weizmann Institute, and U-M. The U-M researchers are led by Betsy Foxman, professor of epidemiology at the School of Public Health. Wiens, who is also a Co-Director of U-M Precision Health, is a Co-PI for the U-M research group.
An accelerated computing platform for genomic sequencing
Blaauw, Das, and Narayanasamy are focused on dramatically accelerating and optimizing the pipeline to process data from the MinION. The goal, say the researchers, is to reduce the time required to analyze a sequenced genome from hundreds of CPU hours to a matter of minutes.
“To realize the full potential of genomic sequencing,” says Das, “computing power needs to increase by orders of magnitude.”
 Enlarge
EnlargeThe problem is, that’s not possible under traditional processor roadmaps, where additional transistors and cores are packed ever more tightly into a processor for incremental processing gains. Added additional programming cores won’t solve the problem either.
“Sustainable growth of processor performance is only possible with custom layers including hardware, software, and algorithms,” says Das.
There are a number of areas of inefficiency that occur during secondary analysis which the team is addressing.
First, says Das, is the read alignment process, during which read data is aligned to genome reference data. Read alignment is composed of two steps: seeding and seed extension.
Seeding finds a set of candidate locations in the reference genome where a read can align. Possible matches are known as hits in the reference. In seed extension, for a read, the reference strings at the hit positions are matched at the read. With current technology, this takes hundreds of CPU hours for a whole genome.
For seeding, the researchers discovered a huge memory bandwidth bottleneck. They did hardware/software codesign and developed a new algorithm, data structure, and index that trades off memory capacity for memory bandwidth. They then built a custom accelerator that traverses the new index efficiently to find hits and seeds. The seeding algorithm has been released as open source software and is planned to be integrated with state of art alignment software from Broad Institute and Intel.
For seed extension, they built a systolic array that would in a few hundred cycles use approximate string matching to match read and reference data.
 Enlarge
EnlargeThe researchers have developed a custom ASIC to eliminate the throughput bottleneck by using a pruning algorithm to optimize the alignment of DNA reads and candidate mutations and by reducing floating point computation by 43x when tested on real human data.
These enhancements and others have been mapped to custom hardware. This includes an accelerator for seed extension which achieves 2.46M reads/second, a ~1800x performance improvement, and a 27x smaller silicon footprint compared to a Xeon E5420 processor.
According to the researchers, when run on a high-end 56-core server in the Amazon cloud, their secondary analysis tools will take about six hours for whole genome sequencing. On an Amazon FPGA server, this reduces to about 20 minutes. When run on the researchers’ custom hardware, processing time is about a minute.
The team has also developed techniques to optimize the read process for pathogen detection. One is to quickly analyze the beginning of a read to determine if it is host or pathogen material. If it is host, the remainder of the read can be skipped since it is only the pathogen material that is of interest. In addition, the researchers are often able to accomplish this host vs. pathogen differentiation using machine learning on squiggle data, without the need for resource-intensive basecalling.
Microbiome analysis to provide faster insights
When processing clinical samples, a fast data processing pipeline is key to the delivery of actionable insights.
“In clinical samples, much of the data — sometimes as much as 90% — can be host DNA, rather than microbial DNA,” says Meera Krishnamoorthy, a PhD student working with Wiens. “As a result, existing metagenomic classification tools store a lot of information about the host, and this can get computationally inefficient.”
 Enlarge
EnlargeIn collaboration with a team of researchers in Michigan Medicine and the School of Public Health, Wiens and Krishnamoorthy are working on in-silico machine learning approaches to host depletion, or the removal of host data reads, which will become a part of Das, Blaauw, and Narayanasamy’s custom hardware. Their goal is to remove all of that host data before classification allowing downstream microbiome analyses to focus solely on microbial data. Existing host depletion methods are laboratory based and can be resource intensive to perform.
In contrast, Krishnamoorthy and Wiens’ approach is computational and does not rely on large reference databases, but instead is based on a convolutional neural network. It takes as input read output by the basecaller and then after a series of convolutions and pooling steps outputs a prediction regarding whether or not the read pertains to the host. The proposed approach proposes to increase the efficiency of downstream analyses, enabling microbiome research that has the potential to transform future medical care.